Step 1 - Create the Species
Draw the Molecule
The first thing we’ll do is draw a benzene molecule as best we can:
Species ⇨ Create ⇨ Draw…
A new editor window opens in which we can draw the rough geometry and connectivity for our molecule.
Select draw mode from the toolbar above the species viewer
Draw a roughly hexagonal ring of six carbon atoms (carbon is the default drawing element) by left-click-dragging in the viewer
Change the drawing element from
CtoHby clicking on the button next to the draw mode icon
Connect a single hydrogen atom to each carbon by left-click-dragging from each carbon site
Your final species will look something like this:You can create bonds by left-click-dragging between two existing atoms.
A very badly drawn benzene molecule
Click to close the editor and create the new species
Double-click on the
NewSpecies tab and change its name to
Apply a Forcefield
Time to make it a little prettier! We’ll assign a standard forcefield to it, and optimise the geometry:
Species ⇨ Add Forcefield Terms…
From the available forcefields choose
OPLSAA2005/Aromaticsand click
You can filter forcefields by keywords in name and description by using the filter box at the top-right of the forcefield selection controls.
We will use the default Determine atom types for all atoms option to add atom types for every atom in the species, so click
There will be no conflicts with existing atom types as there are no atom types already defined, so click
For the intramolecular terms we want to assign them and reduce to master terms (the default settings) so click to proceed
There will be no conflicts with existing master terms, so click to exit the wizard
Click the icon in the species viewer toolbar to optimise the geometry of the molecule using the forcefield we’ve just applied
We will also get ahead here and edit the master terms to reflect the geometry observed in the experimental data, since the forcefield we’ve applied here doesn’t get things quite right.
Go to the
Forcefield tab, section
In the Bonds table change the equilibrium bond length parameter (eq) of the ‘CA-HA’ bond term from
1.08to1.09Å
Change the equilibrium bond length of the ‘CA-CA’ bond term from
1.40to1.38Å
Create Deuterated Isotopologue
Since some of the experimental data was measured on deuterated benzene, we’ll need to create a suitable C6D6 isotopologue:
Go to the
Click to create a new isotopologue for the species
Change the isotope for the HA atom type from
Natural (bc = -3.739 fm)to2 (bc = 6.671 fm)by clicking on the isotopologue entry and choosing from the drop-down menu
Change the name of the isotopologue to ‘C6D6’ (double-click on its name to do so)
The Natural isotopologue is always available and doesn’t need to be created, but doesn’t appear in the list of user-defined isotopologues. It is also not necessary to create “mixed” isotopologues (e.g. for 50:50 mixtures of H:D) as these are created by mixing individual isotopologues.
Add Analysis Sites
We’ll locate our analysis site at the centre of the benzene ring and give it some axes so that we may calculate orientational / spatial functions around it. The figure below shows the atoms we’ll select to define the origin (purple), x-axis (red) and y-axis (blue). Using these atoms as reference points for our coordinate system will set the XY plane to that of the ring, with the z axis perpendicular to the ring, pointing out of its center.
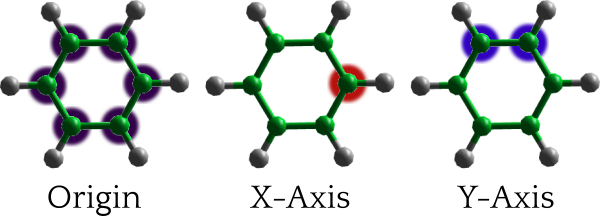
Origin (purple), x-axis (red) and y-axis (blue) atoms defining the oriented benzene site
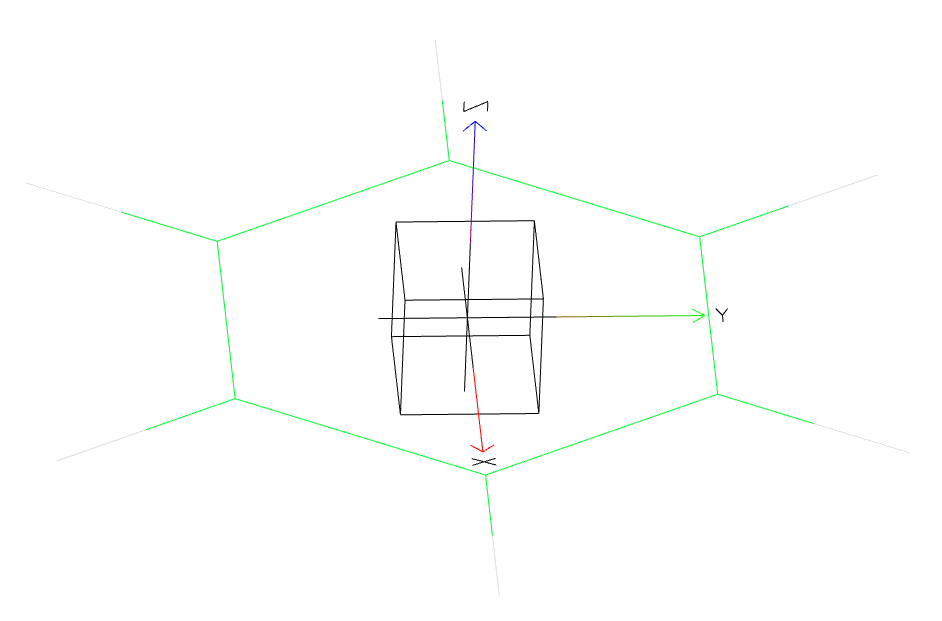
Resulting axis definition in 3D
Go to the
Select all six carbon atoms by shift-clicking on them in the viewer (you may need to click reset view first, to see the whole molecule)
Right-click the selected atoms and click ⇨ This creates a new site with the Carbon atoms as the Origin Atoms
Now select the single carbon atom (as depicted in the image above) and right click it.
Click ⇨ to define the direction of the x axis
Finally, shift-click the pair of adjacent carbon atoms and right click on them
Click ⇨ to define the direction of the y axis
Rename the site to
COGby double-clicking its name