Step 6 - Fix the Water Molecule Geometry
There is a hint in the structure factors for the H2O sample (particularly the G(r)), that the intramolecular geometry of our off-the-shelf forcefield is not quite consistent with the real world. This is made clearly obvious if you look at the G(r) for the D2O sample:
Go to the
G(r) / Neutron S(Q) tab
Click on the “D2O”
NeutronSQ module and select the Output button
Click the button on the “H2O”
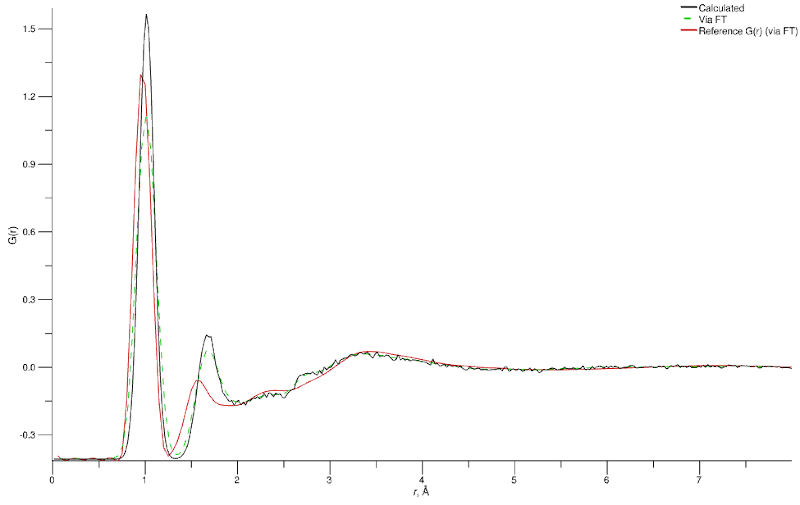
Simulated (black), Fourier-transformed from simulated F(Q) (green), and experimental (red) G(r) for the equilibrated water (D2O) simulation
Clearly we have a mismatch between the experimental (red) and simulated (green) peak positions at around 1 Å (related to the O-H bond) and 1.7 Å (related to the H-O-H angle). It is usually necessary to adjust the geometry of your species a little in order to be consistent with the experimentally-measured data, and in the present case we are lucky that we only have two parameters to adjust!
Here we are modifying the intramolecular terms based on comparison of the D2O data, but bear in mind that liquid water is amongst the systems most sensitive to isotopic substitution since all hydrogens are hydroxyl hydrogens, and subject to exchange as well as strong hydrogen bonding. As such, the differences in intramolecular geometry between H2O and D2O are measurable.[1]
Since we set up our simulation to use intramolecular master terms (via the Add Forcefield Terms… wizard) we can modify those to directly affect our water molecule’s geometry. For the O–H bond it is quite straightforward to read of the true distance (0.976 Å) from the reference g(r). The angle distance requires a touch more trigonometry but, given knowledge of the correct O–H distance, we can work out that the corresponding equilibrium angle we require is 107.134 °.
First of all, let’s stop the simulation from running:
…or…Simulation ⇨ Stop
Esc
And now let’s make the changes to our intramolecular terms:
Go to the
Forcefield tab and find the tab
In the Bonds section, change the equilibrium bond length (’eq’) of the
HW-OWbond term from 1.0 to 0.976 Å
In the Angles section the equilibrium angle (’eq’) of the
HW-OW-HWangle term from 113.24 to 107.134 °
Now run the simulation for a little longer to let the species adjust to their new geometry:
Ctrl-R
You should see a marked improvement in the comparison of the D2O total G(r) and structure factor.
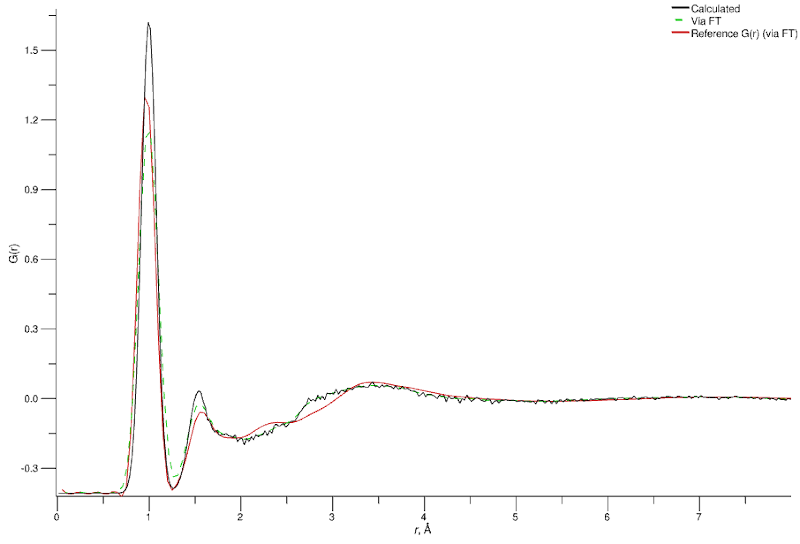
Simulated (black), Fourier-transformed from simulated F(Q) (green), and experimental (red) G(r) for the equilibrated water (D2O) simulation with adjusted intramolecular parameters
The change in the G(r) will not be instant as the majority of the evolution of the system is from the
MolShake module which does not change the intramolecular geometry. Only the
MD module will affect the intramolecular geometry. Also, the g(r) calculated by the
GR are averaged over five calculations by default.
It’s also worth checking the other two samples, where the same kind of improvement should be noticeable (if a little less prominent).
References
- Quantum Differences between Heavy and Light Water, A. K. Soper and C. J. Benmore, Phys. Rev. Lett. 101, 065502 (2008).