Atom Types
Overview
Atom types provide interaction parameters which describe how the energy (and forces) between atoms that are not bound together interact with each other, as a function of distance. You will often hear these referred to as “short-range”, “van der Waals”, or (in specific cases) “Lennard-Jones” interactions. The latter is actually just one popular functional form utilised to represent short-range interactions. These parameters essentially describe how atoms or molecules “feel” each other in a condensed phase simulation, typically preventing overlap of atoms due to their strongly repulsive nature at very short distances, and also contributing a little to cohesive (i.e. negative) energy between molecules. Interaction parameters are specific to one atom type alone - interactions between different atom types are generated automatically by applying “combination rules”.
Atom types will typically be introduced into your simulation through the application of a forcefield on to a species. The total number of atom types in the simulation defines many other things, for instance how many partials result from calculating
 GR">RDFs or
GR">RDFs or
 NeutronSQ">NeutronSQ. Because of this, adding or removing atom types to a simulation which has already been run will likely fail in a lot of nasty ways.
NeutronSQ">NeutronSQ. Because of this, adding or removing atom types to a simulation which has already been run will likely fail in a lot of nasty ways.
The exact relationship between the number of defined atom types $M$ and the number of distinct interatomic potentials (or partials) is $M(M+1) / 2$ - this progression is illustrated in the figure below.
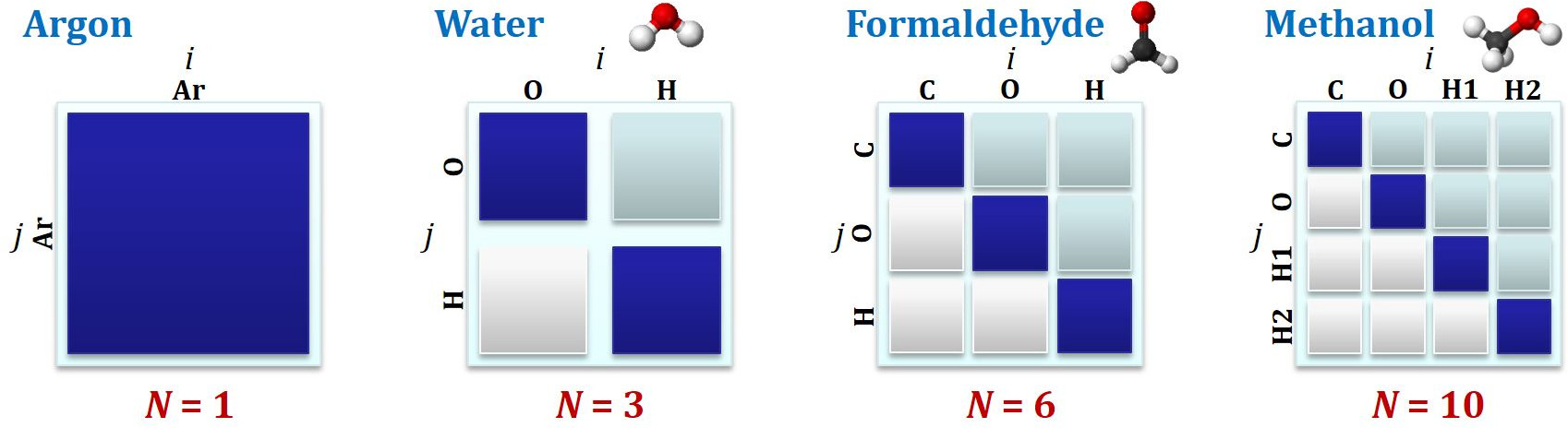
Interatomic potential matrices for simple species
Squares in dark blue (on the diagonal of the matrix) correspond to interactions between similar atom types, while the light blue represent those between different atom types. Squares in white can be neglected because of the equivalence of interaction $i$-$j$ and $j$-$i$. As you can see, even for simple molecules there are a reasonable number of interaction potentials / partials that must be considered, and which rise quickly as more species are added.
Specificity of Atom Types
One key thing to consider with atom types in your simulation is how they affect various processes involving potential refinement, particularly when doing EPSR-like simulations. The number of distinct interatomic potentials resulting from the $M$ atom types present in the system can be thought of as the maximum “flexibility” that potential refinement methods have to work with. Having too few atom types in the system may hamper such methods as there are not enough variable parameters to change. Similarly, having too many atom types (in the extreme case one per individual atom, regardless of chemical similarity of those atoms) can provide too many variable parameters compared to the density of other available information. Usually, published forcefields are sensible enough to reduce atom types down based on molecular symmetry and chemical similarity, but this is something to be aware of if you are constructing your own.
But how can one have “too few” atom types in the system? In a molecule like water of course there can only be two, one shared by the hydrogens and one for the oxygen, but what about a linear alkane such as hexane? Here, it is possible to have just two atom types (one shared by the carbon atoms, and one shared by the hydrogens) or one could quite sensibly partition up the carbons and their attached hydrogens into three atom types each according to their relative positions from each end of the molecule, giving $M = 6$ and 21 distinct interatomic potentials. Partitioning in this way may be helpful in some cases - for the hexane example, if there are distinct end-to-end correlations in the system then separating out the interactions for those terminal carbons may be a sensible thing to do.