Main Forcefield
The purpose of the Forcefield tab is to provide access to general forcefield terms used within the simulation, covering atom types and master terms.
Atom Types
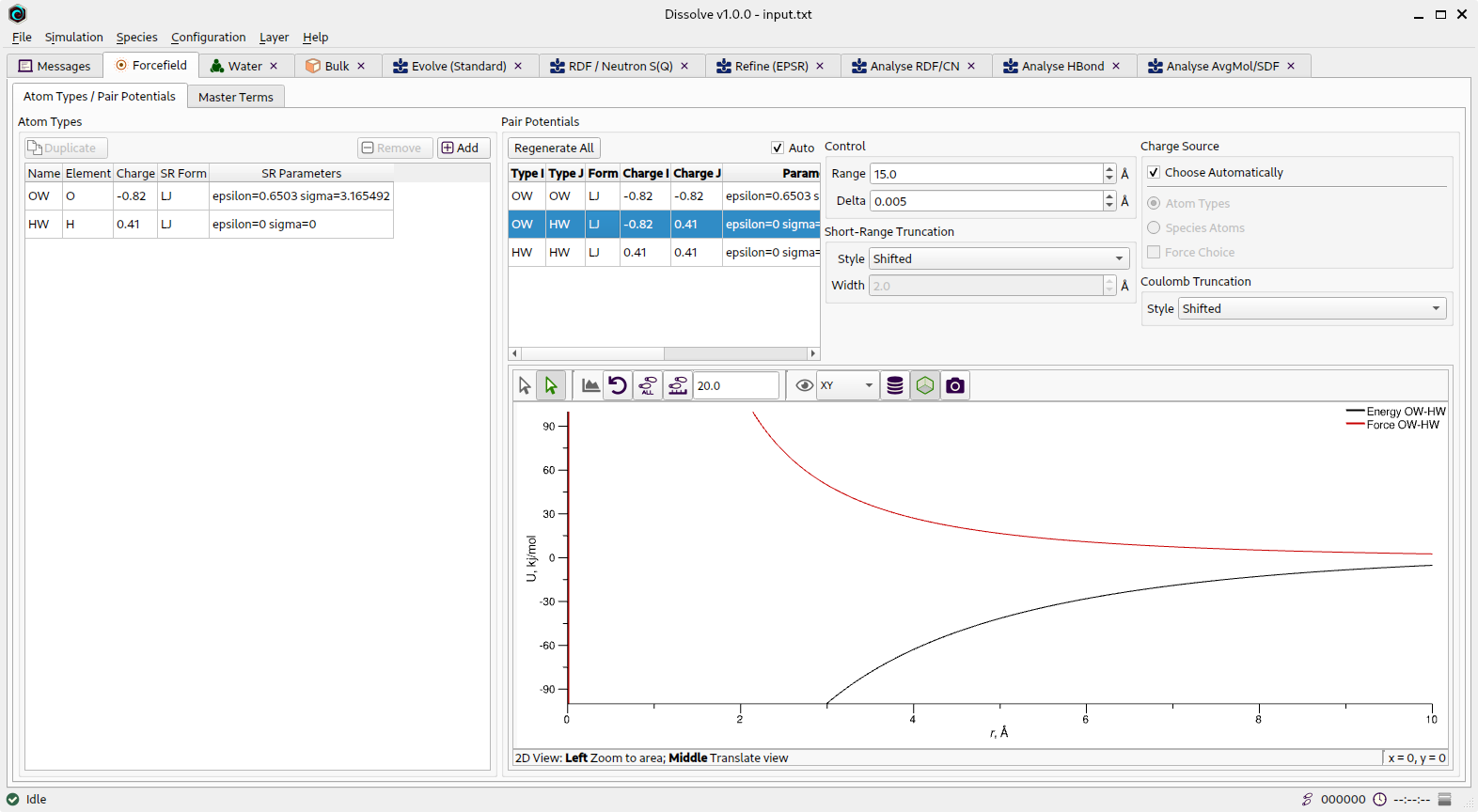
Forcefield tab showing the current atom typess
A full list of all current atom types is given here, including their charges (if using atom types as the charge source - see Pair Potentials below) and short-range (SR) parameters. Everything except the element to which the atom type relates may be freely edited from here. Pair potentials are generated from the atom types listed here automatically by Dissolve unless specifically requested not to.
Pair Potentials
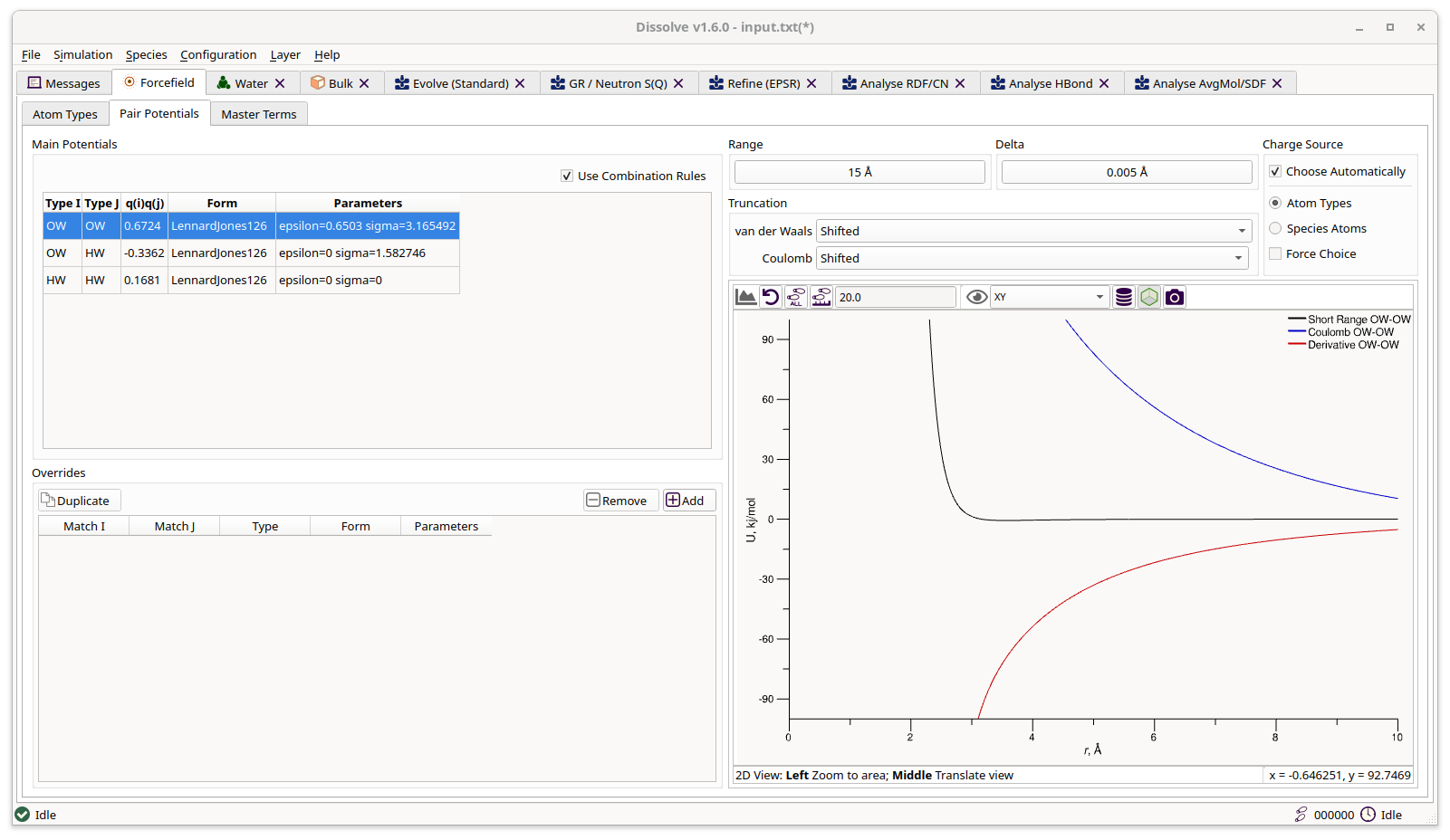
Forcefield tab showing defined pair potentials
Main Potentials
Pair potentials are, by default, generated (tabulated) from the short-range parameters defined for each atom type. This may or may not include the charges defined for individual atom types, depending on the choice of Charge Source (see below). Selecting a pair potential from the list plots the tabulated energy and force functions.
The Use Combination Rules option is checked by default and requests that Dissolve automatically generate all necessary pair potentials when required. This can be un-checked, however, if a custom set of pair potentials is required - for instance, the default set can be generated by the built-in combination rules and then freely edited by unchecking this option.
Overrides
Pair potential overrides are applied after main potentials have been generated and can be used to adjust a potential with a specific additional feature or completely rewrite specific potentials. For example, one may wish to add a repulsive term at shorter distances for one or two potentials, or it might be desirable to completely overwrite a single potential with a new one gleaned from literature, bypassing the one generated by any combination rules.
Pair potential ovverides act upon atom type matches rather than one pair of specific atom types. In other words, you can name a pair of atom types explicitly (e.g. “HW” and “OW” in a water potential) or use wildcards (*) to capture more than one, or even all, atom types. To define an override which should be applied between all pair potentials involving hydrogen and carbon atom types one could use “H*” and “C*” for instance.
Range
The pair potential range determines the extent over which pair potential interactions between atoms are effective in the simulation. Typically this is between 10 to 15 Angstroms, but may need to be made larger in extreme cases. It is important to note that the longer the pair potential range, the longer it takes to calculate total system energies, and the more time it will take your simulation to run.
The Delta value controls the spacing between points, in Angstroms, in the tabulated pair potentials. The default value is sufficient for most cases and should not normally need to be changed.
Truncation
The short-range part of the potential typically goes close to zero at longer distances (past 10 Angstroms, for example) but doesn’t go exactly to zero. This can create a discontinuity in both energy and force which causes significant artifacts in any simulation. Thus, a suitable truncation scheme must be employed to ensure that the energy and force go smoothly to zero at the limit of the pair potential. In an analogous manner to the short-range part of the potential, the Coulombic term must also be made to go to zero at the limit of the pair potential. The “Shifted” scheme is the default and recommended choice.
See also short-range truncation schemes and Coulomb truncation schemes.
Charge Source
When Choose Automatically is enabled for the charge source (the default) Dissolve will make a choice over where to take atom charges for the pair potentials:
- Atom Types - Tabulated pair potentials will include a Coulombic term based on the charges defined for individual atom types in the left-most list.
- Species Atoms - Tabulated pair potentials will only include the short-range part of the potential - Coulomb interactions will be calculated on-the-fly using charges specified on individual species atoms.
Of the two options, Species Atoms is much more flexible in terms of specifying fine-grained, realistic charge distributions, but is slower to calculate.
When attempting to choose which scheme to use Dissolve may fail to make a definite choice and refuse to proceed. This can happen if there are a suspicious number of atom types or species atoms with exactly zero charge, for instance. In those cases you must intervene and untick Choose Automatically and make the choice yourself. Note that Dissolve may still complain about your choice and refuse to run, in which case you must evaluate the situation and, if things are as you want them, select the Force Choice option.
Master Terms
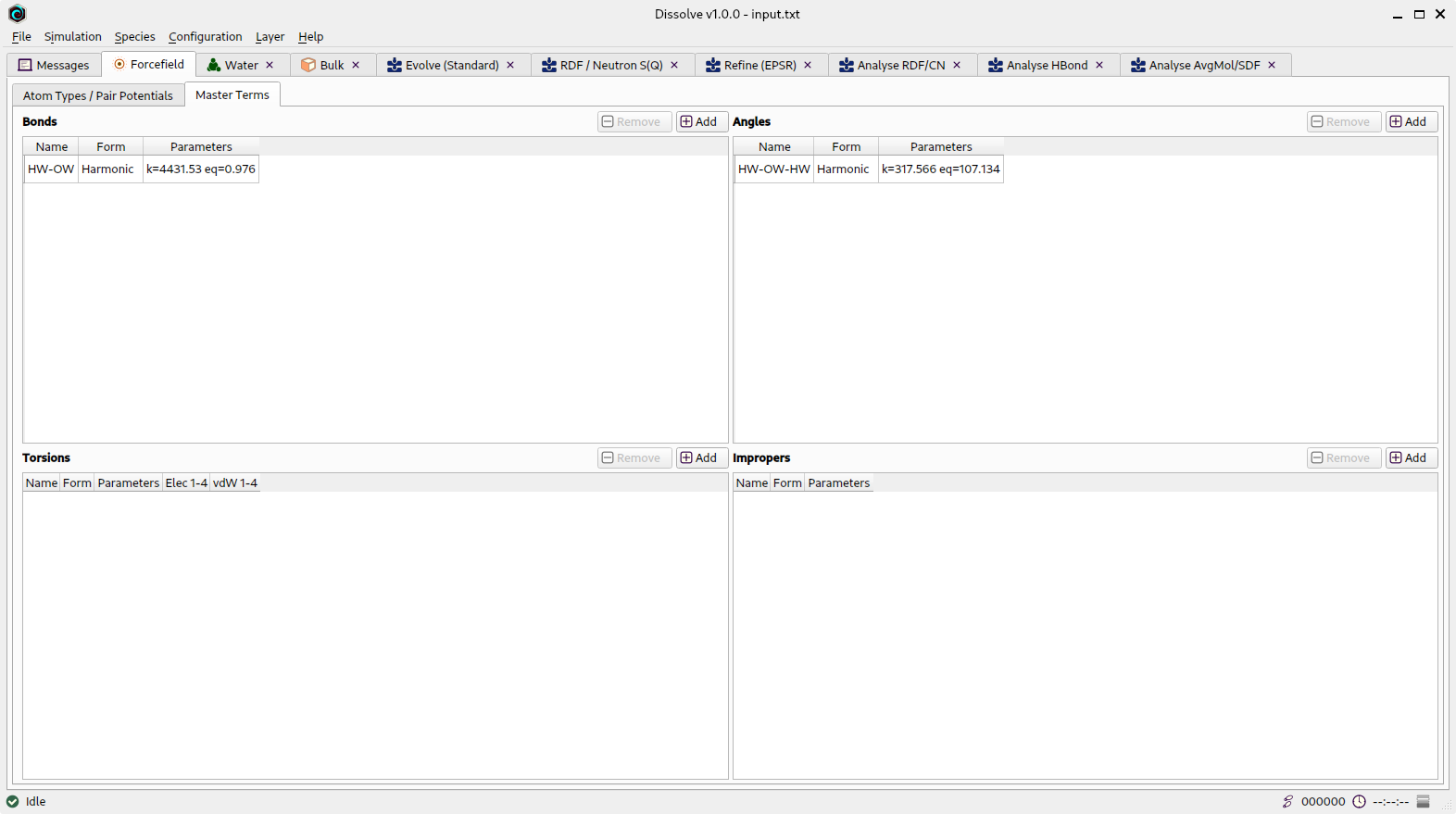
Forcefield tab showing current master terms
Any defined master terms are shown here with their parameters, split into the different intramolecular interaction types. Note that master terms can only be edited from this tab - they cannot be edited from the individual species that employ them (they will show up there as unmodifiable parameters).